
1. Introduzione
La malattia di Fabry (Fabry Disease, FD) è una rara patologia da accumulo lisosomiale, a trasmissione recessiva legata al cromosoma X, causata da mutazioni del gene della galattosidasi-α (GLA), che comportano un’assenza o una riduzione dell’attività dell’enzima α-galattosidasi A (α-AGAL). La carenza di tale enzima determina un progressivo accumulo all’interno dei lisosomi di glicosfingolipidi, quali il globotriaosilceramide (Gb3) e la sua forma deacilata, la globotriaosilsfingosina (lyso-Gb3), con conseguenti alterazioni funzionali cellulari.1 Il progressivo accumulo intracellulare di Gb3 è responsabile dell’attivazione di pathways pro-infiammatori, responsabili a loro volta di indurre processi pro-fibrotici, contribuendo allo sviluppo di una malattia multi-sistemica con danno a carico di organi vitali come cuore, rene e cervello.2
Infatti, pur configurandosi come una malattia sistemica, gli esiti clinici principali sono riconducibili soprattutto al progressivo interessamento dell’apparato renale, cardiovascolare e del sistema nervoso centrale.1
Il coinvolgimento cardiaco, caratterizzato principalmente dalla cardiomiopatia ipertrofica e dalla comparsa di aritmie cardiache, rappresenta uno dei principali determinanti prognostici negativi. Del resto, anche la progressione dell’insufficienza renale ed il verificarsi di eventi cerebrovascolari ricorrenti condizionano in modo significativo la qualità di vita (QoL) e l’aspettativa di vita dei pazienti.3
Oltre alle principali manifestazioni a carico di cuore, rene e sistema nervoso centrale, la FD si associa a un ampio spettro di altre manifestazioni multi-sistemiche, che contribuiscono in modo significativo al burden clinico complessivo della malattia.4,5 (Figura 1)
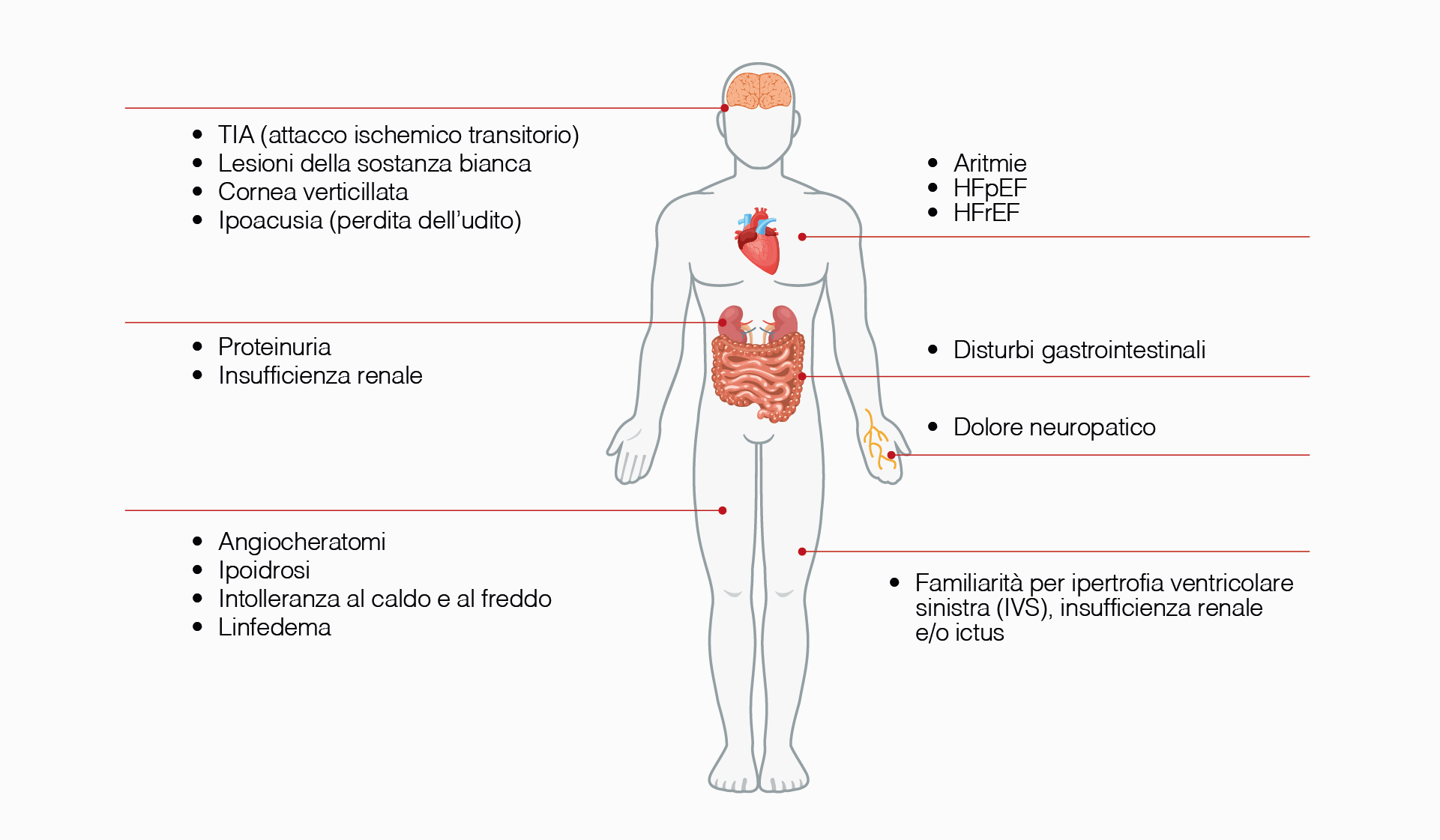 Figura 1. Principali manifestazioni cliniche della malattia di Fabry.
Figura 1. Principali manifestazioni cliniche della malattia di Fabry.
Coinvolgimento multisistemico della FD con interessamento del sistema nervoso centrale e periferico, dell’apparato cardiovascolare, renale, gastrointestinale, cutaneo e sensoriale.
Dal punto di vista epidemiologico, la prevalenza della FD è stimata tra 1 caso ogni 40.000-170.000 nati vivi.6 Tali stime sono state messe in discussione da studi di screening neonatale e in gruppi ad alto rischio, che riportano frequenze decisamente più elevate e confermano come la patologia rimanga tuttora largamente sottodiagnosticata a causa della sua eterogeneità clinica.7 La malattia può essere suddivisa in un fenotipo classico severo in assenza di attività enzimatica residua e in un fenotipo non classico generalmente più lieve.
La FD non classica, definita anche a esordio tardivo o atipica, è caratterizzata da un decorso clinico variabile, nella quale i pazienti presentano generalmente un minore coinvolgimento sistemico, di minore severità con manifestazioni di malattia che possono essere limitate a un singolo organo.8
I pazienti di sesso maschile, spesso caratterizzati da un fenotipo classico a causa di un’attività dell’enzima GLA assente o minima, sviluppano tipicamente sintomi a esordio precoce, a differenza dei maschi con forma non classica, caratterizzata da un’attività residua di GLA con un quadro clinico più lieve ed a esordio tardivo.9 Al contrario, le pazienti di sesso femminile eterozigoti per mutazioni del gene GLA possono presentare un ampio spettro di manifestazioni cliniche o rimanere completamente asintomatiche.9
Questa diversità di quadri clinici dipende dal fatto che l’inattivazione del cromosoma X avviene con modalità asimmetriche (skewed inactivation), ovvero la percentuale del cromosoma con il gene mutato può essere diversa, nelle singole pazienti, rispetto al cromosoma sano, contribuendo a determinare il fenotipo.10
La terapia enzimatica sostitutiva (Enzyme Replacement Therapy, ERT), disponibile da oltre vent’anni, ha rappresentato un punto di svolta nella gestione dei pazienti affetti da FD, contribuendo in modo sostanziale a migliorare l’evoluzione clinica e la prognosi dei pazienti. Attualmente sono disponibili formulazioni enzimatiche somministrate per infusione endovenosa a cadenza quindicinale. Si distinguono, infatti, l’agalsidasi alfa, prodotta in linee cellulari umane, e somministrata alla dose di 0,2 mg/kg di peso corporeo e l’agalsidasi beta, prodotta in colture cellulari di ovaio di criceto cinese e impiegata alla dose di 1 mg/kg di peso corporeo.11
Più recentemente è disponibile una ulteriore formulazione enzimatica quale la pegunigalsidasi-alfa, costituita da due molecole alfa legate in maniera covalente associate polietilenglicole (PEG), e prodotta utilizzando cellule vegetali di tabacco. Oltre alla ERT, per i pazienti con FD è disponibile la terapia orale “chaperonica” con Migalastat sebbene limitata ai pazienti portatori di mutazioni suscettibili, mentre studi clinici di terapia genica sono attualmente in corso.12-14
È opportuno ricordare che in assenza di trattamento, l’aspettativa di vita nelle donne e negli uomini risulta ridotta rispettivamente di circa 10 e 20 anni.15 Per analizzare le caratteristiche della FD, Axenso ECM, con il contributo non condizionante di Takeda, ha sviluppato nel 2025 il Progetto FORWARD Fabry Outcomes, Research, Work & Advanced Real-world, un percorso comunicazionale ECM strutturato, articolato in due eventi, uno residenziale e uno online.
L’iniziativa ha coinvolto un gruppo multidisciplinare di 22 esperti italiani afferenti alle aree della cardiologia, nefrologia, medicina interna e immunoallergologia, con l’obiettivo di fornire un inquadramento complessivo e integrato della gestione della malattia, in linea con la natura multi-sistemica della FD. Nel corso degli eventi è stata condotta una survey finalizzata ad ottenere una fotografia aggiornata della gestione della FD in diversi Centri italiani; inoltre, è stato realizzato un lavoro a gruppi mediante la metodologia “metaplan” ed è stato richiesto ai partecipanti di condividere e descrivere casi clinici rappresentativi. Il confronto si è concentrato, in particolare, sugli aspetti fisiopatologici emergenti, sulle implicazioni immunologiche della ERT e su specifiche criticità organizzative del percorso diagnostico-terapeutico.
Il presente articolo si propone di delineare una sintesi ragionata delle evidenze cliniche, fisiopatologiche e organizzative emerse dal confronto multidisciplinare, focalizzandosi sulle principali criticità e sulle aree a maggiore impatto per l’ottimizzazione della gestione della FD. Tali tematiche sono state integrate da un’analisi critica della letteratura scientifica di riferimento, al fine di contestualizzare le evidenze emerse e rafforzarne l’interpretazione alla luce dei dati più recenti disponibili.
2. Il ruolo dell’agalopatia e le altre vie patogenetiche nella progressione del danno d’organo nella malattia di Fabry
La patogenesi della FD è tradizionalmente attribuita al deficit di α-AGAL con conseguente accumulo lisosomiale di Gb3 e compromissione delle funzioni lisosomiali stesse. Tuttavia, evidenze crescenti indicano che ulteriori fattori patogenetici possano contribuire allo sviluppo e alla progressione della malattia.16-20
Sebbene l’accumulo lisosomiale di Gb3 rappresenti un evento patogenetico centrale, non appare sufficiente, da solo, a spiegare la complessità e l’eterogeneità delle manifestazioni cliniche osservate nei pazienti. L’accumulo intracellulare di Gb3 attiva, infatti una rete di pathways interconnessi – tra cui infiammazione, stress ossidativo, disfunzione mitocondriale ed endoteliale, alterazione dell’autofagia e del metabolismo energetico – che contribuiscono in modo autonomo e progressivo al danno d’organo.21,22 (Figura 2)
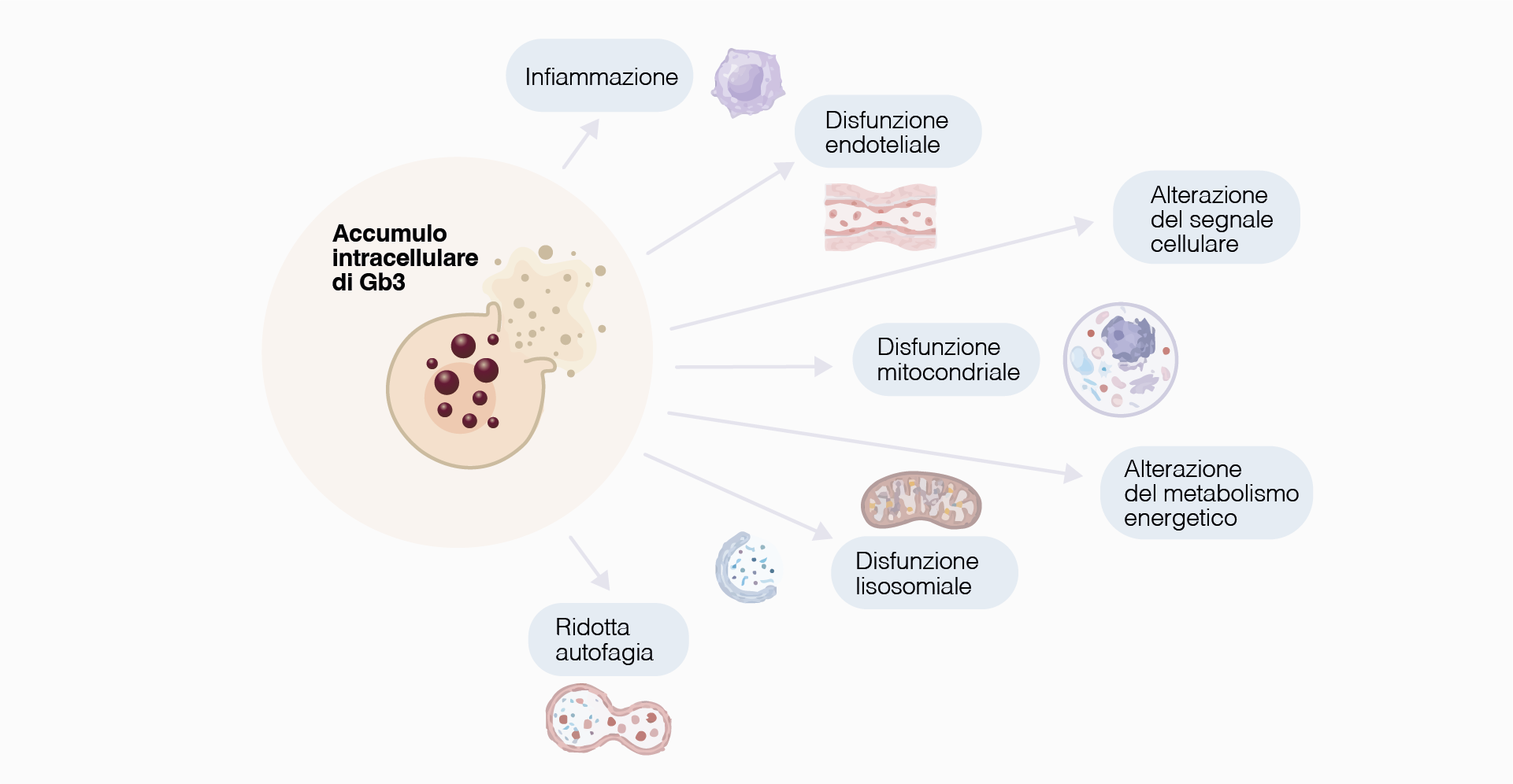 Figura 2. Rappresentazione schematica dei principali meccanismi cellulari coinvolti nella FD.
Figura 2. Rappresentazione schematica dei principali meccanismi cellulari coinvolti nella FD.
Figura rielaborata da Thomson SE, 202522.
Nel corso del Progetto FORWARD è stato discusso il concetto di agalopatia, inteso come modello interpretativo di disfunzione dell’α-AGAL associata ad alterazioni della proteostasi cellulare. In linea con quanto recentemente descritto in letteratura, alcune varianti missenso dell’α-AGAL possono determinare un “misfolding” proteico e ritenzione intracellulare nel reticolo endoplasmatico della proteina stessa, con conseguente attivazione dello stress del reticolo endoplasmatico e della unfolded protein response (UPR), fenomeni che possono contribuire a processi di infiammazione, apoptosi e disfunzione cellulare.2
Tali osservazioni hanno portato alla proposta del concetto di “AGALopathy”, secondo cui il danno cellulare in corso di FD deriverebbe non solo dal deficit enzimatico e dall’accumulo lisosomiale di Gb3, ma anche da un’alterazione primaria della proteostasi legata alla ritenzione e al “misprocessing” della proteina mutata.23 Questo modello suggerisce l’esistenza di meccanismi patogenetici almeno in parte indipendenti dall’accumulo lisosomiale, e potenzialmente rilevanti soprattutto nelle forme non classiche della malattia. È da considerare anche che una parte della proteina mutata può sfuggire al sistema reticolo-endoteliale ed essere immessa in circolo seppur in quantità ridotta. Pertanto, i meccanismi patogenetici associati alla UPR possono, in alcune mutazioni, coesistere con le alterazioni dovute alla deposizione di lyso-Gb3.23
In ambito nefrologico, il podocita rappresenta un bersaglio cruciale del danno nella FD. Evidenze istopatologiche hanno dimostrato che la frazione del citoplasma podocitario occupata da Gb3 aumenta progressivamente con l’età, raggiungendo un plateau relativamente precoce; tuttavia, tale stabilizzazione del carico lisosomiale non si accompagna a un arresto del danno cellulare. Al contrario, i marcatori di stress podocitario e la perdita di podociti continuano a progredire nel tempo, con comparsa di podocituria e successiva evoluzione della nefropatia.24
Un ulteriore meccanismo patogenetico è rappresentato dal danno tubulo-interstiziale. La deposizione di Gb3 determina nelle cellule epiteliali la trasformazione in cellule mesenchimali, ovvero miofibroblasti. Queste cellule hanno la capacità di secernere citochine sia infiammatorie che fibrosanti e possono inoltre migrare sino ai vasi e ai glomeruli. Il risultato finale è la progressiva fibrosi del tessuto renale. Nello studio delle biopsie renali dei pazienti Fabry non è inusuale osservare quadri istologici caratterizzati prevalentemente da componenti infiammatorie e fibrotiche tubulo-interstiziali, non sempre accompagnate dai reperti glomerulari significativi.25
Tali evidenze sui possibili meccanismi patogenetici di danno renale sono in accordo con l’ipotesi di meccanismi di danno non esclusivamente substrato-dipendenti e possono contribuire a spiegare alcune difficoltà diagnostiche.4
Questi dati suggeriscono che, nelle fasi più avanzate della malattia, meccanismi patogenetici diversi dal solo accumulo di Gb3 possano contribuire in modo significativo alla progressione del danno d’organo. Ciò evidenzia l’importanza di una comprensione integrata dei pathways coinvolti, al fine di migliorare l’inquadramento clinico e la gestione complessiva della patologia.
L’importanza dei meccanismi di stress cellulare è stata ulteriormente rafforzata dalla discussione multidisciplinare emersa nel corso della sessione mattutina, nella quale è stata sottolineata la persistente difficoltà nel definire una correlazione genotipo–fenotipo univoca in una malattia clinicamente così eterogenea. È stato sottolineato come la fisiopatologia della FD non possa essere ricondotta esclusivamente all’accumulo lisosomiale di Gb3 o alla semplice instabilità enzimatica, ma coinvolga l’attivazione di pathways patogenetici ulteriori, in parte condivisi con altre patologie microvascolari cerebrali. Tali meccanismi potrebbero contribuire a spiegare alcuni aspetti neurologici ancora non completamente chiariti. Nel complesso, è emersa la necessità di superare una visione esclusivamente “storage-centrica” della malattia, riconoscendone la natura fisiopatologica più complessa e multifattoriale.
3. Gli ADA nella terapia della malattia di Fabry
Un tema ampiamente dibattuto sia nel corso della sessione dedicata all’immunogenicità e nel successivo confronto multidisciplinare riguarda il ruolo degli anticorpi anti-farmaco (anti-drug antibodies, ADA) nei confronti della ERT. È stato dimostrato che circa il 40-70% dei pazienti maschi affetti da FD nella forma classica sviluppa anticorpi IgG anti-α-galattosidasi A durante il trattamento con ERT; la formazione di ADA è stata tuttavia descritta anche nelle pazienti di sesso femminile e in soggetti con forme non classiche della malattia.26 La presenza degli ADA è risultata associata a una ridotta efficacia della ERT con potenziale impatto negativo sugli outcome clinici nei pazienti che li sviluppano.27,28
Gli ADA possono essere distinti in anticorpi neutralizzanti, in grado di interferire con il legame recettoriale e di inibire l’attività enzimatica, ed anticorpi “clearanti”, cioè capaci di aumentare la clearance dell’enzima, con conseguente interferenza sulla farmacocinetica e farmacodinamica dell’enzima. È stato dimostrato che l’inibizione dell’ERT mediata dagli ADA si associa a livelli più elevati di lyso-Gb3, maggiore severità clinica (Mainz Severity Score Index, MSSI) in particolare nei pazienti maschi con FD nei quali la produzione di ADA risulta essere maggiore.27
Il ruolo degli ADA con attività neutralizzante è stato messo in luce anche nello studio di Lenders et al, (J Am Soc Nephrol, 2018) nel quale si dimostrava che dopo otto anni di ERT la riduzione media del lyso-Gb3 risultava strettamente correlata al grado di inibizione anticorpale dell’enzima, con una diminuzione del 25% nei pazienti con minore inibizione rispetto all’8% nei soggetti con maggiore attività neutralizzante degli ADA.28
Del resto, gli ADA neutralizzanti determinano un’inibizione dell’enzima infuso già nel corso dell’infusione, compromettendo sia l’uptake endoteliale dell’enzima sia la sua attività enzimatica a livello intracellulare.28-30
Per quanto concerne il potenziale immunogenico delle ERT è da sottolineare come le modalità di sintesi, in particolare dalla linea cellulare impiegata per l’espressione dell’enzima ricombinante, sia uno dei fattori determinanti. Infatti, sebbene la sequenza primaria aminoacidica di tutte le ERT approvate riproduca fedelmente quella dell’α-galattosidasi A umana nativa, le differenze nei “pattern” di glicosilazione post-traduzionale rappresentano un determinante rilevante del profilo immunogenico delle diverse formulazioni. 26
L’agalsidasi alfa, prodotta in fibroblasti umani, presenta una glicosilazione analoga a quella dell’enzima endogeno; l’agalsidasi beta, ottenuta da cellule ovariche di criceto cinese, contiene residui di acido N-glicolilneuraminico (NGNA), una sialilazione diversa da ciò che è presente sulla proteina nativa umana condizionate un “folding” diverso e nel complesso un potenziale maggiore di immunogenicità.26 Anche la pegunigalsidasi alfa può indurre la formazione di ADA, fenomeno in parte correlato sia al profilo di glicosilazione non umano sia alla componente PEG; in un’ampia coorte internazionale di pazienti con FD, la positività agli ADA è stata riportata in circa il 30% dei soggetti trattati.32
Un aspetto clinicamente rilevante è rappresentato dalla cross-reattività degli ADA: anticorpi diretti contro agalsidasi alfa o beta possono riconoscere reciprocamente le due molecole ed anche pegunigalsidasi alfa, seppur con affinità variabile.31-33
Per tale motivo lo switch terapeutico tra le due formulazioni di agalsidasi non garantisce necessariamente il superamento dell’immunogenicità mediata dagli ADA e può determinare risposte eterogenee, includendo in alcuni pazienti un incremento dei titoli anticorpali.31 Tali osservazioni suggeriscono che il cambio di molecola debba essere valutato con cautela e accompagnato da un attento monitoraggio clinico, immunologico e farmacocinetico.
È noto che la formazione di ADA si associa a lo sviluppo di reazioni infusionali (infusion-related reactions, IRRs) oltre alla la riduzione dell’efficacia della ERT, sebbene in una parte dei pazienti che sviluppano ADA non si osservino outcome clinicamente negativi almeno a breve termine.26 Alla luce di queste evidenze, la valutazione immunologica del paziente assume un ruolo sempre più centrale nella gestione della FD, in particolare nei casi di risposta terapeutica subottimale, di peggioramento clinico non chiaro o in previsione di modifiche del trattamento. Risulta essenziale sviluppare strategie volte a limitare la formazione di ADA ed a promuovere quindi una tolleranza immunologica duratura nei confronti dell’enzima, in particolare nei pazienti naïve.26 L’identificazione dei soggetti ad alto rischio di sviluppo di ADA prima dell’inizio della ERT rappresenta un passaggio cruciale per il raggiungimento di questo obiettivo.26
4. La diagnosi precoce: campanelli d’allarme
Alla luce della complessità fisiopatologica descritta e dell’eterogeneità clinica che ne deriva, la diagnosi precoce della FD rappresenta tuttora una sfida clinica rilevante, in considerazione della natura multi-sistemica della patologia e della scarsa specificità dei sintomi nelle fasi iniziali. Manifestazioni precoci quali acroparestesie, crisi dolorose, ipo- o anidrosi, intolleranza al calore e febbre di origine sconosciuta risultano spesso aspecifiche e possono determinare un prolungato iter, con ricorso a molteplici consulenze specialistiche e frequenti diagnosi errate, configurando una vera e propria “odissea diagnostica”.34-37 (Figura 3)
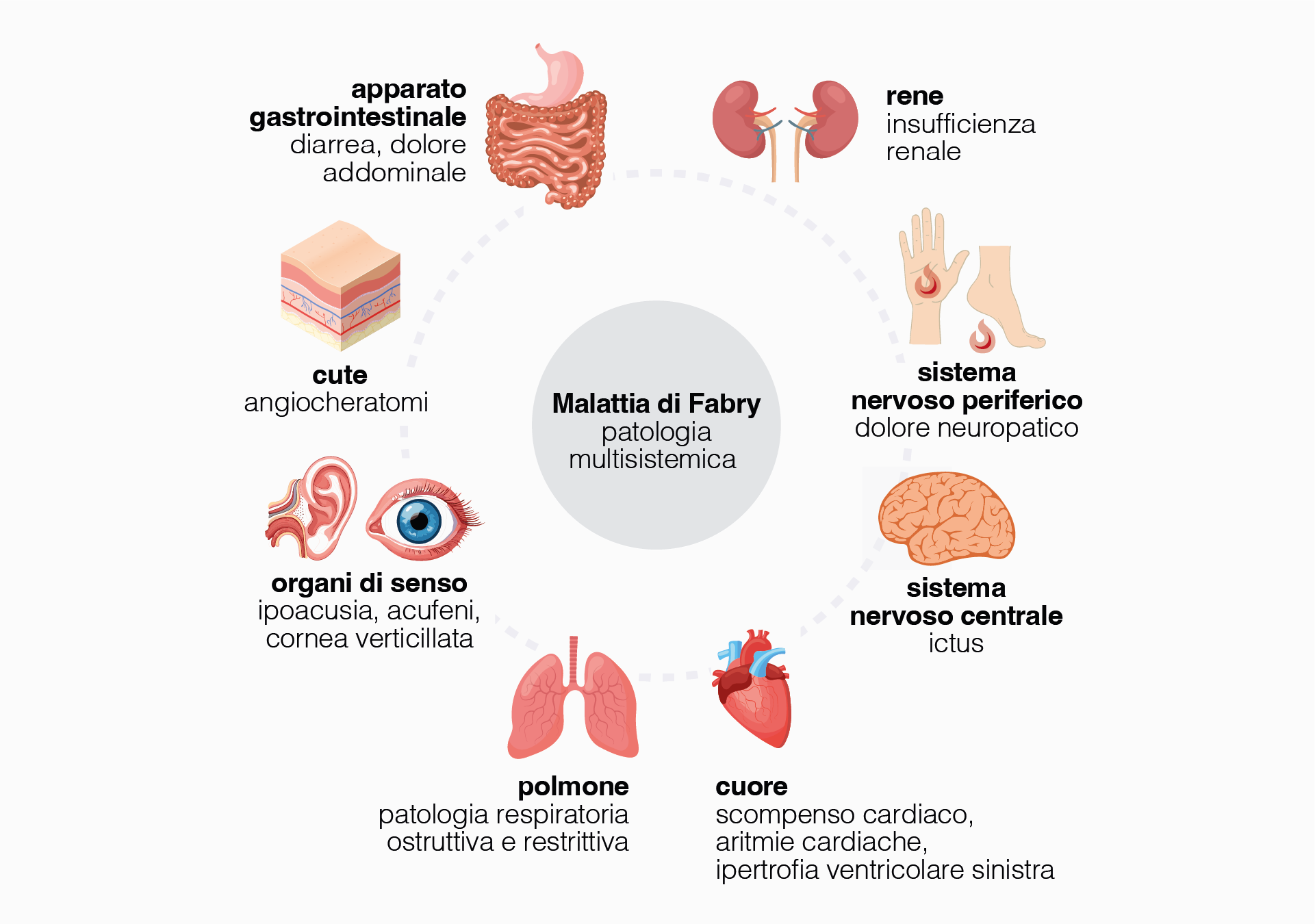 Figura 3. Manifestazioni cliniche multisistemiche della FD.
Figura 3. Manifestazioni cliniche multisistemiche della FD.
Rappresentazione schematica dei principali organi e apparati coinvolti nella FD e delle relative manifestazioni cliniche associate. Figura rielaborata da Lenders M, Brand E, 202238.
Un ulteriore elemento di complessità è rappresentato dal coinvolgimento differenziale dello stesso organo nelle diverse fasi di malattia, rendendo difficile l’identificazione precoce della FD.39
In ambito cardiologico, le manifestazioni precoci possono includere alterazioni elettrocardiografiche che precedono lo sviluppo di anomalie strutturali evidenti, quali intervallo PR corto, anomalie della ripolarizzazione e segni iniziali di ipertrofia ventricolare sinistra. Dal punto di vista strumentale, l’ecocardiografia con studio della meccanica cardiaca tramite strain può evidenziare segni precoci, prima della comparsa dell’anomalia cardiaca strutturale più comune che è l‘ipertrofia ventricolare sinistra mentre la risonanza magnetica cardiaca consente la caratterizzazione tissutale che può rivelare bassi valori di T1 mapping come primo fenomeno.40-41
Con la progressione della malattia compaiono reperti più avanzati, quali elevati voltaggi del QRS, “pattern di strain”, inversione profonda dell’onda T e disturbi della conduzione, IVS e fibrosi.39-42
L’eterogeneità clinica della FD richiede pertanto un approccio personalizzato nella gestione del paziente, che tenga conto del genotipo, del sesso, della storia familiare, del fenotipo e della specifica severità delle manifestazioni cliniche di ciascun individuo.39 Un numero crescente di evidenze indica che l’avvio precoce della ERT è associato ad una migliore risposta clinica. Nei pazienti adulti di sesso maschile con mutazione classica, l’inizio tempestivo del trattamento, anche in assenza di sintomi evidenti, può contribuire a prevenire la progressione verso danni tissutali irreversibili e insufficienza d’organo.
Alla luce di queste considerazioni, risulta fondamentale un maggiore coinvolgimento dei pediatri e dei medici di medicina generale nel sospetto diagnostico della malattia, nonché una formazione mirata degli specialisti finalizzata al riconoscimento delle manifestazioni cliniche specifiche della FD nei diversi ambiti di competenza.
5. Vantaggi della terapia domiciliare
La complessità clinica della FD e la necessità di un trattamento enzimatico sostitutivo continuativo nel tempo rendono fondamentale considerare non solo l’efficacia biologica della terapia, ma anche la sua sostenibilità organizzativa e il suo impatto sulla qualità di vita.
La ERT ha dimostrato di determinare benefici clinici rilevanti nella FD; tuttavia, la necessità di somministrazioni endovenose” long-life”, con cadenza quindicinale, può interferire con le attività della vita quotidiana e determinare un impatto negativo sulla QoL dei pazienti. La disponibilità di programmi di terapia domiciliare rappresenta una modalità sicura e praticabile almeno in una parte dei pazienti, associata ad un miglioramento dell’esperienza terapeutica e a una riduzione del carico assistenziale complessivo. (Figura 4)
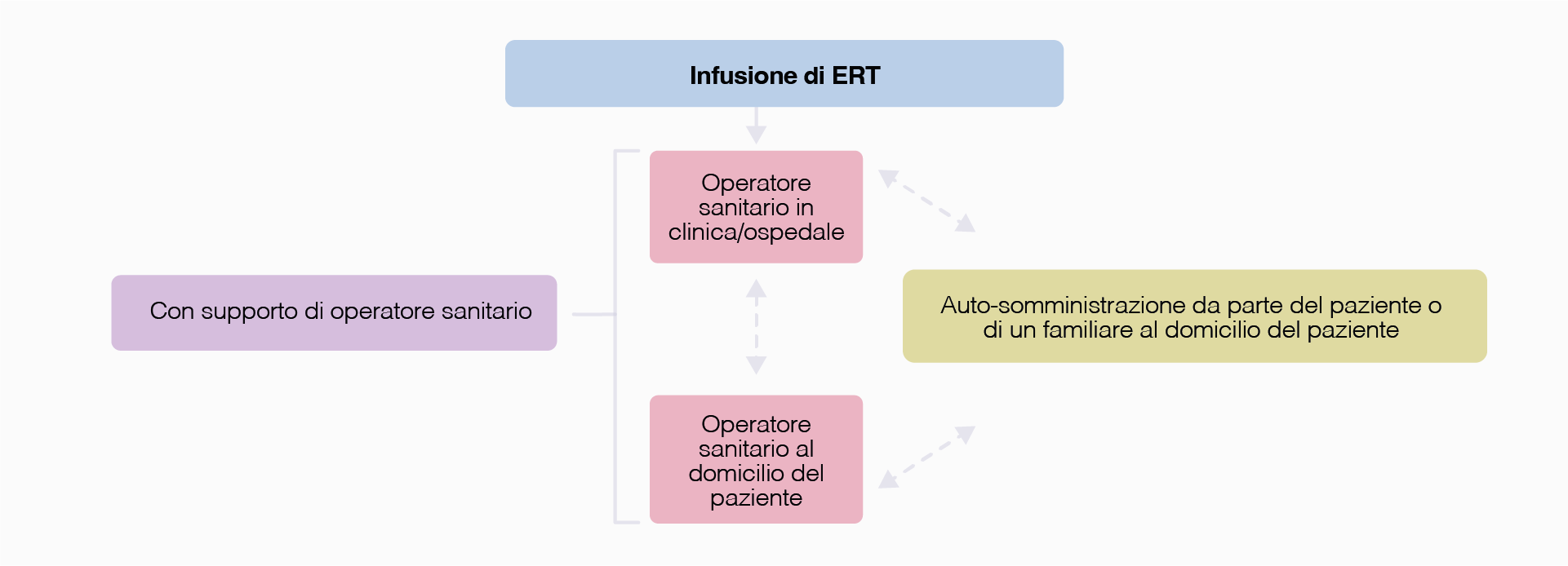 Figura 4. Transizione tra modalità di ERT.
Figura 4. Transizione tra modalità di ERT.
I pazienti possono passare in modo flessibile dall’ERT somministrata da operatori sanitari all’auto-somministrazione domiciliare, con possibilità di ritorno temporaneo al setting clinico in caso di eventi avversi.
Dati recenti indicano che la transizione dall’infusione domiciliare assistita all’auto-somministrazione non comporta un aumento degli eventi avversi severi rispetto alla somministrazione da parte di operatori sanitari.43 Nella coorte europea proveniente dai registri FOS/GOS, sebbene di non facile spiegazione, l’incidenza di eventi avversi e di IRR è risultata inferiore nel gruppo in self-administration rispetto a quello trattato con supporto sanitario, mentre gli eventi severi sono risultati sovrapponibili tra i due gruppi.38 Inoltre, la terapia domiciliare risulta associata a un miglioramento dell’aderenza terapeutica e a un effetto significativo e favorevole sulla QoL.44,45
Lo studio di Concolino et al. (2017) descrive l’esperienza di un ampio gruppo collaborativo italiano sull’impiego della ERT a domicilio nei pazienti affetti da FD e rappresenta una delle più ampie evidenze disponibili su questo modello terapeutico.46 L’analisi, osservazionale e longitudinale, ha coinvolto 85 pazienti (45 maschi e 40 femmine) provenienti da 11 Regioni italiane, per un totale di 4.269 infusioni domiciliari di agalsidase-alfa nell’ambito del programma Fabry@Home. I risultati hanno mostrato un’elevata applicabilità del modello domiciliare, con un’aderenza terapeutica pari al 100%, un miglioramento o una stabilità degli indicatori di QoL nella maggior parte dei pazienti e una stabilità del punteggio MSSI, indice validato di severità clinica della FD, che integra manifestazioni generali, neurologiche, cardiovascolari e renali). Tale andamento si è distinto rispetto al lieve peggioramento osservato nel precedente periodo di gestione ospedaliera. Gli eventi avversi sono risultati rari, di lieve entità e gestibili a domicilio, a conferma della sicurezza del modello nei pazienti adeguatamente selezionati.46
Nel complesso, la terapia domiciliare con ERT si configura come un modello assistenziale strutturato, associato a benefici clinici e organizzativi documentati, con potenziali implicazioni favorevoli anche in termini di sostenibilità sanitaria. In Italia, tali programmi possono essere attivati attraverso l’Assistenza Domiciliare Integrata (ADI) o mediante programmi domiciliari gestiti da agenzie esterne, spesso con il supporto delle aziende produttrici. Il percorso prevede un’accurata selezione del paziente, un’adeguata informazione e acquisizione del consenso da parte dello stesso, un periodo iniziale di addestramento in ambiente ospedaliero in assenza di reazioni infusionali o eventi avversi clinicamente rilevanti, seguiti dall’avvio del trattamento a domicilio. (Figura 5)
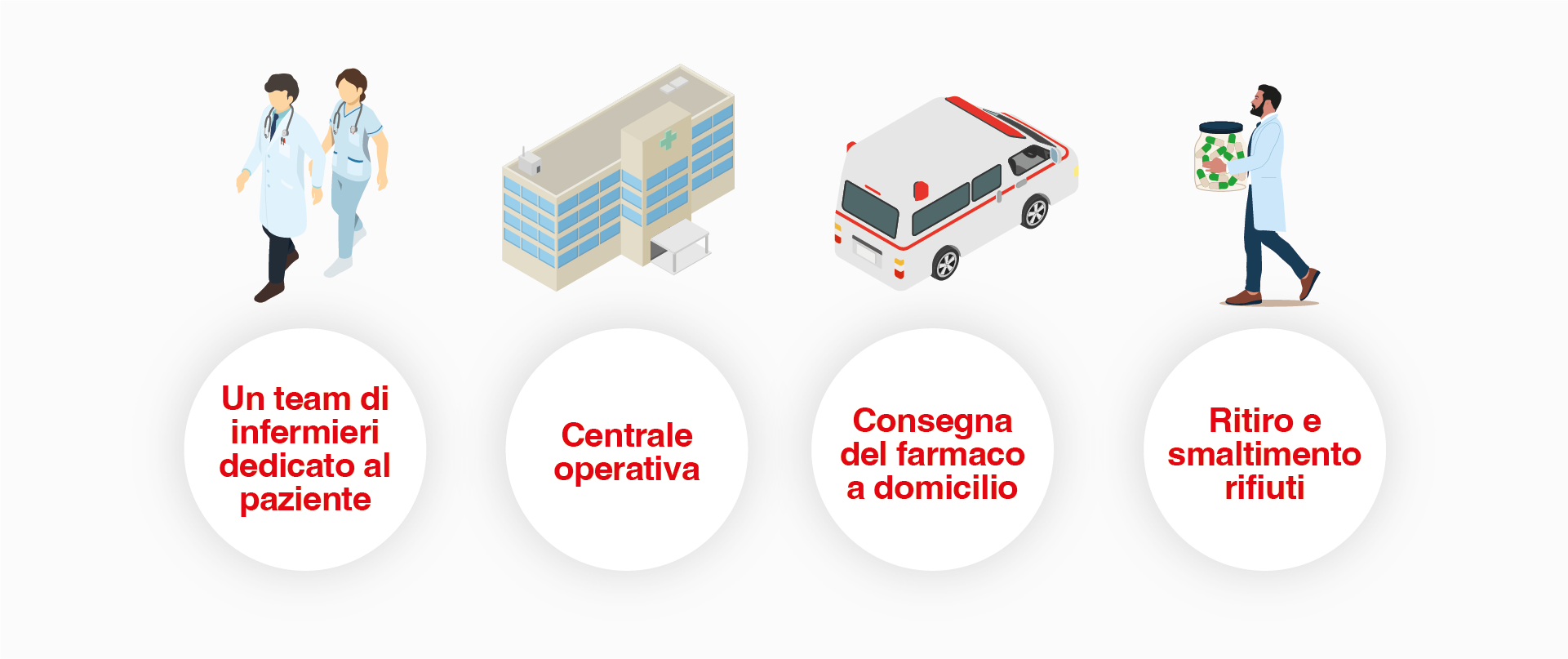 Figura 5. Vantaggi della terapia domiciliare.
Figura 5. Vantaggi della terapia domiciliare.
Dal punto di vista del paziente, la terapia domiciliare consente una maggiore adattabilità del trattamento alla vita quotidiana senza la necessità di recarsi in ospedale con un conseguente miglioramento della QoL, peraltro, in condizioni di sicurezza e con un incremento dell’aderenza terapeutica. L’esperienza maturata durante la pandemia da Covid-19 ha permesso di confermare l’utilità della ERT domiciliare nel garantire continuità terapeutica, con un miglioramento della percezione di sicurezza da parte dei pazienti.
Sotto il profilo economico, l’analisi condotta da Heinrich et al. ha evidenziato che i costi della ERT domiciliare sono come atteso prevalentemente determinati dal costo del farmaco, ma vi è un risparmio della quota relativa al mancato utilizzo delle risorse sanitarie per il personale medico ed infermieristico ed all’uso delle strutture ospedaliere. Pertanto, il trasferimento del trattamento al domicilio non risulta associato a un incremento dei costi rispetto alla somministrazione in ambiente ospedaliero.47
6. Survey Fabry Care: dalla diagnosi alla presa in carico. Quali criticità superare?
I partecipanti al Progetto FORWARD, afferenti a diversi centri distribuiti sul territorio nazionale, hanno preso parte alla survey dal titolo “Fabry Care: dalla diagnosi alla presa in carico. Quali criticità superare?”. Il campione era costituito da 21 specialisti provenienti da differenti ambiti disciplinari, a testimonianza della natura multi-sistemica della FD e della conseguente necessità di un approccio multidisciplinare nella gestione clinica. (Figura 6)
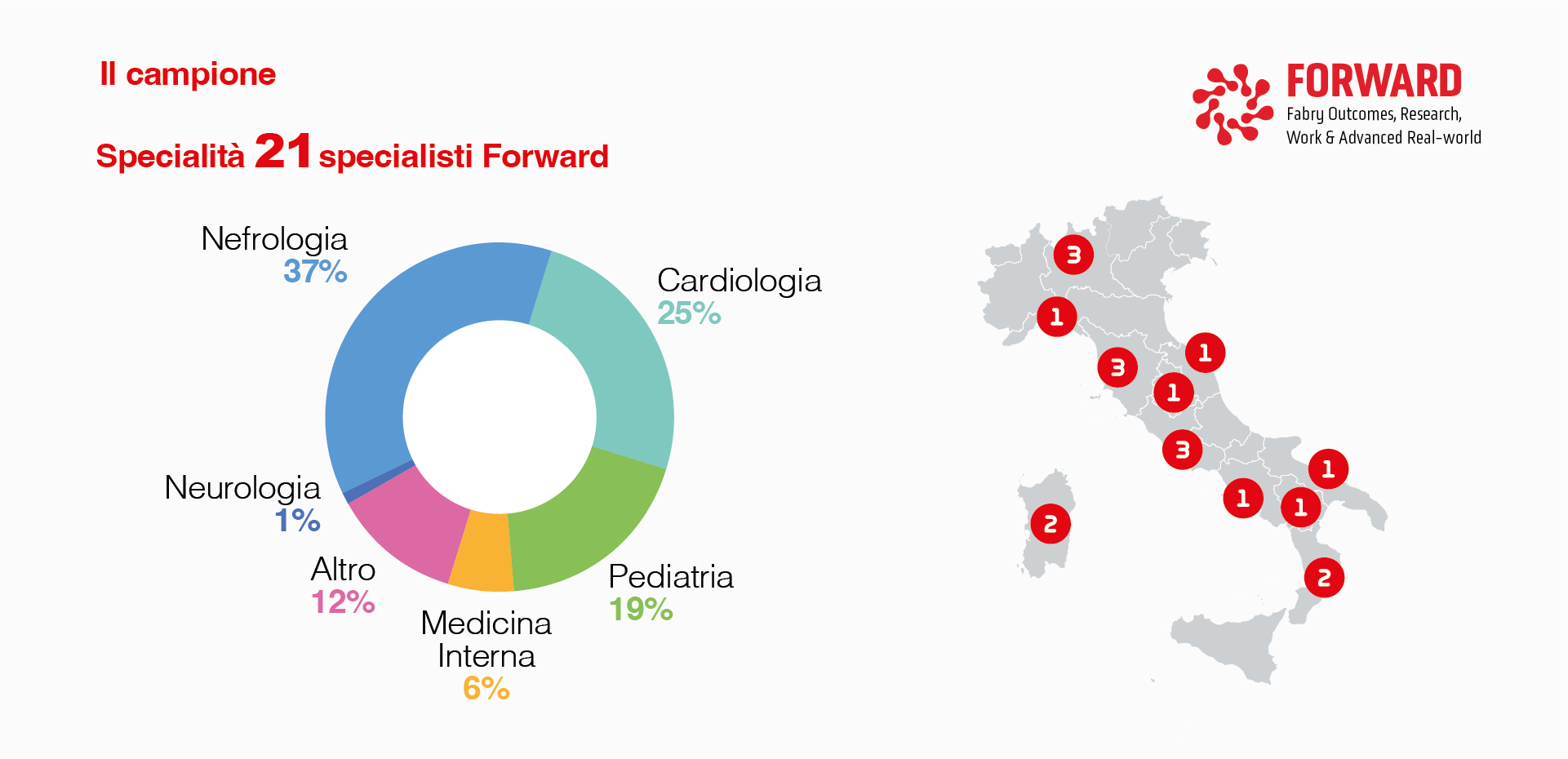 Figura 6. Distribuzione per specialità e area geografica dei partecipanti alla survey del Progetto FORWARD.
Figura 6. Distribuzione per specialità e area geografica dei partecipanti alla survey del Progetto FORWARD.
L’indagine mirava a raccogliere l’esperienza diretta degli esperti coinvolti nella gestione dei pazienti con FD, al fine di confrontare le pratiche adottate nei diversi centri italiani e identificare criticità e punti di forza nel percorso di diagnosi, presa in carico e follow-up.
La survey ha evidenziato una gestione multidisciplinare non uniformemente strutturata, con percorsi formalizzati solo in circa la metà dei Centri e un coordinamento tra specialisti percepito come pienamente efficace in una quota minoritaria di casi. La comunicazione tra specialisti e medico di medicina generale risulta poco strutturata, essendo giudicata pienamente efficace solo nel 7% dei casi.
Nel follow-up, il 50% dei clinici segnala difficoltà di accesso ai controlli periodici e discontinuità assistenziale, mentre nel 30% dei casi emergono problematiche di comunicazione tra specialisti. Gli aspetti psicologici e motivazionali sono indicati come il principale fattore che incide sull’aderenza terapeutica (26%), seguiti da elementi organizzativi quali la durata delle infusioni e la distanza dal centro di somministrazione.
Analizzando i fattori che condizionano complessivamente il percorso diagnostico-terapeutico nella FD, la diagnosi precoce emerge come priorità principale (78,6%), seguita dal coordinamento tra specialisti (57,1%) e dal supporto psicologico (50,0%). Aderenza terapeutica ed educazione del paziente sono considerate rilevanti nel 35,7% dei casi, mentre l’accesso alle terapie è indicato con minore frequenza (14,3%).
Nel complesso, i risultati dell’indagine rafforzano la necessità di modelli di presa in carico maggiormente strutturati, fondati su un coordinamento multidisciplinare efficace, sull’educazione del paziente e sull’adozione di strumenti organizzativi condivisi (quali PDTA e figure di Case Manager), al fine di migliorare la continuità assistenziale e l’aderenza nel lungo periodo. (Figura 7)
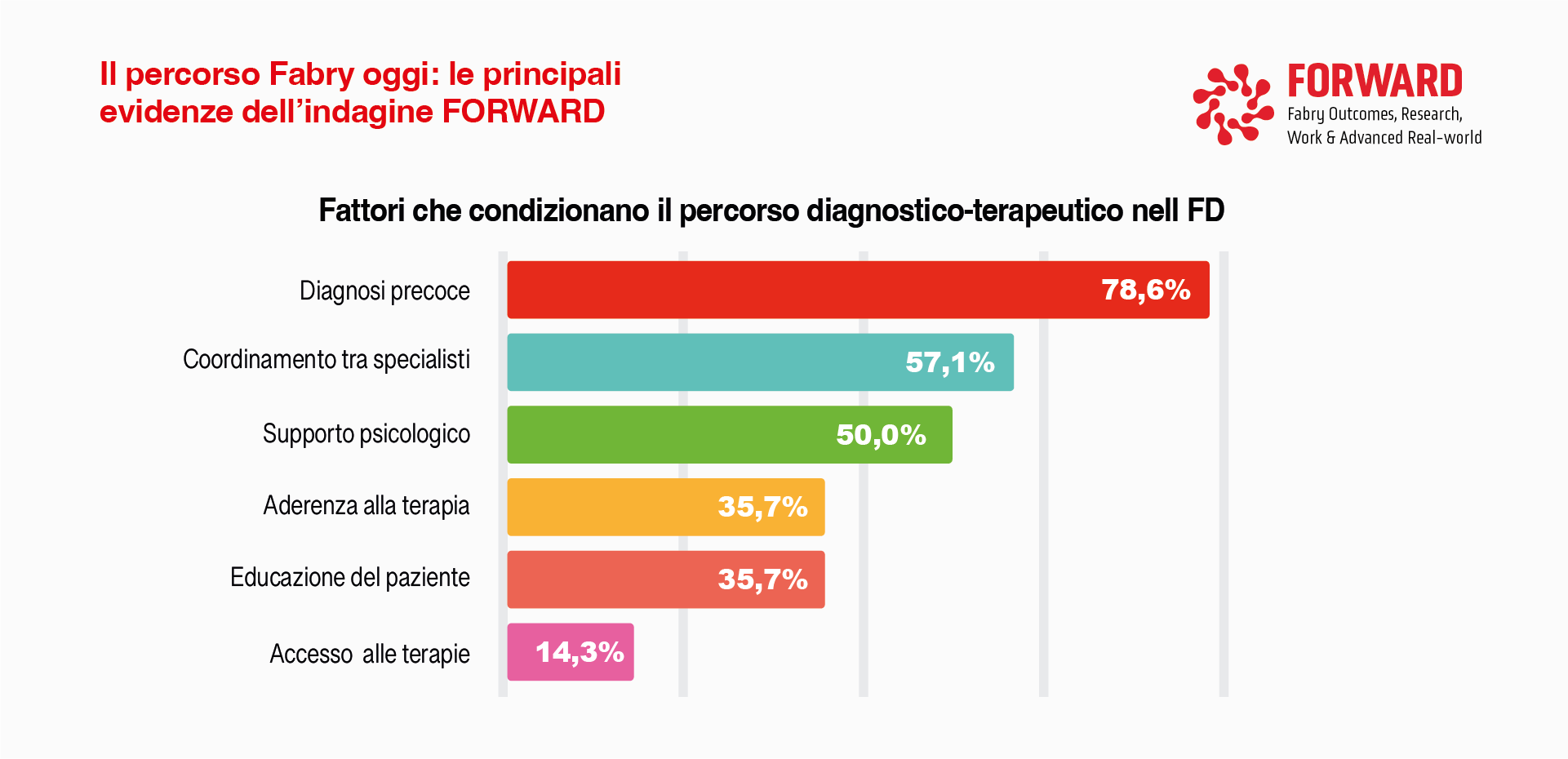 Figura 7. Fattori prioritari nel percorso diagnostico-terapeutico della FD secondo la survey FORWARD.
Figura 7. Fattori prioritari nel percorso diagnostico-terapeutico della FD secondo la survey FORWARD.
Conclusioni
Il Progetto FORWARD (Fabry Outcomes, Research, Work & Advanced Real-world) si è configurato come un percorso strutturato di confronto multidisciplinare, finalizzato a integrare evidenze scientifiche aggiornate, esperienza clinica reale e analisi dei modelli organizzativi adottati nei diversi Centri italiani. Attraverso sessioni tematiche dedicate agli aspetti fisiopatologici, terapeutici e gestionali della malattia, l’iniziativa ha consentito di mettere in relazione la crescente complessità biologica della FD con le criticità operative della presa in carico, favorendo un’analisi integrata dei determinanti clinici e organizzativi degli esiti a lungo termine.
La malattia di Fabry è una patologia complessa la cui gestione richiede un approccio integrato che superi una visione esclusivamente “storage-centrica” e tenga conto dei molteplici meccanismi patogenetici coinvolti, inclusi quelli legati alla disfunzione della proteostasi e all’immunogenicità della terapia enzimatica sostitutiva. L’evoluzione delle conoscenze fisiopatologiche, unitamente alla disponibilità di diverse opzioni terapeutiche, impone una valutazione personalizzata del paziente, fondata sull’integrazione di dati clinici, biochimici, genetici e strumentali.
L’immunogenicità della ERT, con particolare riferimento allo sviluppo di ADA, rappresenta un determinante rilevante della risposta terapeutica e sottolinea la necessità di un monitoraggio strutturato e di strategie orientate alla prevenzione e alla gestione degli ADA, soprattutto nei pazienti ad alto rischio. Accanto agli aspetti strettamente clinici, rivestono un ruolo determinante l’assetto organizzativo dei percorsi assistenziali e la qualità del coordinamento multidisciplinare. La tempestività della diagnosi, l’integrazione strutturata tra specialisti e medico di medicina generale, l’attenzione ai determinanti psicologici dell’aderenza terapeutica e l’implementazione di percorsi formalizzati di presa in carico costituiscono elementi essenziali per garantire continuità assistenziale e ottimizzare l’efficacia complessiva del percorso di cura.
La terapia domiciliare, nei pazienti selezionati, si configura come un’opzione sicura e sostenibile, in grado di migliorare l’esperienza terapeutica e favorire la continuità assistenziale, senza incremento dei costi complessivi. L’esperienza del Progetto FORWARD ha offerto un’opportunità di confronto strutturato tra specialisti provenienti da diversi Centri italiani, consentendo di integrare evidenze scientifiche, pratica clinica reale e analisi organizzativa. I risultati emersi confermano l’esigenza di rafforzare i modelli multidisciplinari e di promuovere strumenti condivisi di presa in carico, in un’ottica di miglioramento continuo della qualità assistenziale.
Nel complesso, emerge la necessità di un modello di presa in carico dinamico e realmente multidisciplinare, capace di adattarsi nel tempo all’evoluzione clinica del singolo paziente e di integrare innovazioni terapeutiche, monitoraggio immunologico e strumenti organizzativi condivisi, con l’obiettivo di preservare la funzione d’organo e migliorare gli outcomes a lungo termine.
Elenco componenti Gruppo FORWARD
- Egrina Dervishi, Firenze
- Teresa Faga, Catanzaro
- Maurizio Gallieni, Milano
- Alessia Gennarini, Bergamo
- Rosita Greco, Cosenza
- Andrea Guido, Genova
- Maria Angela Losi, Napoli
- Stefania Marazia, Lecce
- Giacomo Mascia, Cagliari
- Davide Noto, Palermo
- Claudia Sgattoni, Ancona
- Ludovico Luca Sicignano, Roma
- Irene Sitzia, Cagliari
- Costantino Smaldone, Potenza
- Martina Tedesco, Brescia
- Antonino Tuttolomondo, Palermo
- Cinzia Zuchi, Perugia
Iniziativa realizzata grazie ad un contributo non condizionante di ![]()

Complimenti per questa messa a punto di una malattia ancora sottodiagnosticata, dai risvolti fisiopatologici complessi e ancora in parte inesplorati. L'aver istituito il progetto Forward rappresenta una vera e propria svolta che porterà all'istituzione di PDTA della malattia che attraverso la condivisione multidisciplinare consentirà l'effettiva presa in carica del paziente. La conoscenza dell'immunogenicità della terapia consentirà inoltre una personalizzazione della stessa e un ulteriore avvicinamento a una medicina di precisione